Introduction
The increase in body fat and decrease in muscle mass during aging are associated with physical deterioration and degenerative diseases and cause obesity via the accumulation of abdominal fat. This increases insulin resistance as well as the production of cytokines (tumor necrosis factor-α, TNF-α; interleukin-4, IL-4; monocyte chemoattractant protein-1, MCP-1), which cause inflammation, and adipocytokines, which promote catabolism in muscles, thereby reducing muscle/myo-function. These processes lead to obesity and ultimately to insulin resistance syndrome, diabetes, and cardiovascular diseases. Considering the recent domestic trend characterized by a rapidly aging population owing to an extension in the average life span and change in the population composition, it is necessary to analyze sarcopenic obesity and insulin resistance and to develop strategies for active prevention.
In this review, the effects of aerobic exercise training, one of the major preventative methods for obesity, on endoplasmic reticulum (ER) stress and sarcopenic obesity caused by obesity and aging as well as insulin resistance syndrome were determined to analyze the correlations between these phenotypes.
ER stress and unfolded protein response
The ER is an organelle found in cells of all eukaryotes and consists of tubules, vesicles, and cisterna in a netlike form. It is composed of smooth ER, with a membranous structure that extends from the nuclear membrane, and rough ER with attached ribosomes. ER functions in protein synthesis, folding, modification and transport to different cells [1]; approximately one-third of all intracellular proteins are produced within the rough ER. The smooth ER is important for lipid and sterol synthesis, calcium storage, and the regulation of the intracellular calcium concentration [2]. Immature proteins can be introduced to the ER in various physiological or pathological conditions, resulting in ER dysfunction due to the depletion of calcium; this ER dysfunction is referred to as ER stress [3]. The abnormal protein aggregation such as misfolded and unfolded protein result in an increase proteotoxicity [4].
The cellular response to ER stress is well known as unfold protein response (UPR) which seeks to recover ER function. UPR has multiple strategies to restore ER homeostasis. UPR increases ER chaperone proteins to inhibit protein aggregation and facilitate correct protein folding, and UPR also reduces protein translation by temporary. Moreover, ER increases own volume by stimulating the synthesis of membrane lipids. Finally, endoplasmic reticulum-associated protein degradation (ERAD) increases a degradation of unfolded proteins [5]. Several ER stress sensors are involved, any increased misfolded and unfolded protein is detected by those sensors.
The sensors of UPR is composed by three branches including inositol-requiring enzyme-1 (IRE1), PKRlike ER kinase (PERK), and activating transcription factor 6 (ATF6). IRE1 mediates the unconventional splicing of the mRNA encoding X-box binding protein-1 (XBP1) [6,7]. XBP1 is a potent trans-activator that regulates genes involved in ER protein synthesis, folding, glycosylation, ERAD, redox metabolism, autophagy, lipid biogenesis and vesicular trafficking [8]. The capacity of IRE1 to sense ER stress that is depends on its dissociation from binding immunoglobulin protein (Bip)/glucose regulated protein 78 (GRP78) and on its interaction with unfolded protein [9]. Both of B-cell lymphoma 2 (Bcl-2)-assoicated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak), pro-apoptotic Bcl-2 family members are essential for IRE1α activation [10]. IRE1 also involves distinct stress pathways including JNK and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-кB) [4].
The second ER stress sensor is PERK. BiP/GRP78 in bound to the PERK luminal domain, when UPR has not been activated. If unfolded protein accumulation is increased, then BiP/GRP78 dissociates from PERK. The overexpression of BiP/GRP78 attenuates this activation [11]. PERK activation induces the phosphorylation of the eukaryotic translation initiation factor 2 alpha (eIF2a), which inhibits translation by decreasing protein influx into the ER [12]. As such, PERK pathway reduces the ER load. But, some mRNAs including transcription factor 4 (ATF4) are translated, leading to the upregulation of genes related to redox homeostasis, amino acid metabolism, autophagy, and apoptosis control [13]. Moreover, previous study has been shown that PERK activation modulates the activity of nuclear factor erythroid 2related factor 2 (NRF2) and forkhead box O (FOXO), both pathway involve the antioxidant response, insulin responsiveness, and autophagy [14]. When ER homeostasis is restored, activated PERK is dephosphorylated [11]. Also, active eIF2α is dephosphorylated by two phosphatases, known as CReP [15] and GADD34 [16].
The third ER stress sensor is ATF6 which is transcription factor. Under normal condition, ATF6 is stable by binding the BiP/GRP78 and calreticulin in the ER [17]. ATF6 translocate to the Golgi apparatus in response to ER stress, where it is sequentially cleaved by the site-1 and then Site-2 Proteases (S1P and S2P, respectively) [18]. Then the N-terminal fragment of ATF6, known as p50ATF6, translocate to the nucleus to promotes transcription of UPR gens including BiP/GRP and GRP94 [2,19], the transcription factors C/EBP homologous protein (CHOP) [20] and XBP1 [7] and sarco/endoplasmic reticulum Ca₂+-ATPase (SERCA) [21]. ATF6 also regulate ER volume via XBP-1 independent manner, and enhance cellular adaptation to chronic ER stress [22].
Notably, the activity of UPR stress sensor is regulated via the binding of co-factors and post-translational modifications that modulate the amplitude of their downstream signals and the kinetics of activation and attenuation [4]. Under chronic ER stress, the UPR involves the upregulation of CHOP, the induction of oxidative stress, exacerbated regulated IRE1-dependent decay (RIDD), upregulation of pro-apoptotic components of the Bcl-2 family, among other mechanisms to mediate apoptosis via different mechanisms [23]. Therefore, under conditions of ER stress, the UPR reprograms the cell toward adaptation, sustaining cell function or the engagement of cell death programs to eliminate irreversibly damaged cells [24].
UPR and autophagy
Autophagy is a cellular homeostatic process to regulate degradation and recycling of protein and organelle. A well-orchestrated program including over 30 Autophagy related-protein (Atg) genes control autophagy, which can be activated by the UPR as aggregated misfolded proteins accumulate [25].
Emerged evidences have shown that ER stress initiates autophagy. It is well known IRE1α, one of the UPR sensor, mediates the stress kinases Jun N-terminal kinase (JNK) [5], which is a core component in autophagic signaling pathway [26]. JNK mediated Bcl-2 phosphorylation interferes with its binding to the pro-autophagy BH3 domain-containing protein Beclin-1 [27]. In addition, activation of XBP1 by IRE1 also triggers autophagy via transcriptional activation of Beclin-1 [28]. Another UPR sensor PERK is essential for autophagy, ER stress by cellular stress induces PERK activation that triggers a transcription of autophagy gene such as Atg, Becn1 and so on by eIF2α/ATF4 pathway, and induction of CHOP by ATF4 also regulates more than a dozen Atg genes [29].
ER stress & obesity in muscle
Recent studies have focused on the correlations between obesity, insulin sensitivity, and type 2 diabetes and ER stress [30]. Since skeletal muscle accounts for more than 80% of insulin-mediated glucose uptake and fatty acid oxidation or the engagement of cell death prostress and incomplete fatty acid oxidation, known as lipotoxicity, results in an increase insulin resistance in skeletal muscle that causes to the development of insulin resistance in the whole body. Lipotoxicity is related to ER stress in skeletal muscle. And ER stress is involved in the link between inflammation and insulin resistance, both of all contribute to the development of T2DM in skeletal muscle. Indeed, Bip/GRP78 protein expression was increased, and GRP78 and XBP1 mRNA also increased in human biceps brachii muscle from T1DM patient [31]. That paper also showed that phosphorylation of PERK and eIF-2α and expression of caspase-3 and -7 in T1DM patient muscle using immunohistochemistry. Another study has been shown that palmitate exposure increases the mRNA of CHOP, ATF3, XBP1, and the spliced form XPB1s as well as inflammatory response in cultured primary human skeletal muscle cells from biopsies of the vastus lateralis muscle.
Disorders in skeletal muscle fatty acid managing have been identified in the pathology of insulin resistance and T2DM and higher postprandial VLDL-TAG extraction across forearm muscle were concurred with decreased postprandial insulin sensitivity in subjects with impaired glucose metabolism compared with normal glucose tolerance [32]. Moreover, the study of VLDL exposure in skeletal muscle has been shown to increase ER stress marker Bip and phospho-eIF2α, whereas no change was found CHOP. VLDL exposure in muscle also resulted in an increase mRNA of IL-6, CMP1 and TNF-α, as well as IRS Ser307 phosphorylation, and reduced IkBα [33], these data indicate that disruption of fatty acid handling in skeletal muscle can induce ER stress, insulin resistance and inflammation.
Bip/GRP78 enhances the ER protein folding capacity and to reduce the unfolded protein load in the ER, when activated this chaperone, insulin signaling pathway is decreased [34]. In the meantime, CHOP increase in a protein synthesis and oxidation in the ER. Under chronic ER stress, the expression of both Bip/GRP78 and CHOP is increased in obesity. Previous study showed that chronic HFD diet of mice and palmitate treatment in C2C12 cell increases both of GRP78 and CHOP [35]. Tribbles 3 (TRB3) is pseudokinase and contains a kinase domain without enzymatic activity. TRB3 is expressed in various tissues such as skeletal muscle, heart, liver and adipose tissue. TRB3 is an inducible factor in the ER stress pathway. ER stress caused by induction of TRB3 in cardiomyocyte inhibits Akt activation. And TRB3 overexpression in C2C12 has been shown to inhibit insulin-stimulated Akt phosphorylation and decreases insulin-stimulated glucose uptake. Previous study has shown that high 70% fat diet result in an increase BiP/GRP78, IRE1α and membrane-bound transcription factor protease site2 (MBTPS2), this protease involved in the ER stress response, protein in mouse soleus. However, UPR response in tibialis anterior on high fat diet was less than soleus, author suggested about the difference UPR between muscle types could be due to greater susceptibility of oxidative fibers to a high fat diet [36]. Another possibility in the differential UPR response between muscle types is that resistance on ER stress in type I muscle is higher than type II. UPR function is exist to reduce over-loaded in ER, type I muscle is higher tolerance on insulin resistance than type II. Interestingly, High fat fed mice for 6 week resulted in an increase cleaved caspase/caspase-12 ratio in tibialis anterior, but the ratio in soleus is no change [36].
Disruption of redox balance in ER induces ER stress, which increases reactive oxygen species (ROS) production in the ER and mitochondria [37]. ROS are considered as second messengers between ER stress and mitochondria, although it is not clear that whether ER stress increases mitochondrial dysfunction or mitochondrial malfunction induces ER stress. Previous study has shown that high fat diet increases oxidative stress and mitochondrial dysfunction as well as ubiquitination of protein, which is maker of both ubiquitin-proteasome and autophagic degradation of protein, in muscle tissue, these data are linked with an increased PERK phosphorylation and JNK [38], indicating that high fat diet induced oxidative stress and mitochondrial dysfunction in muscle is associated with ER stress and autophagic protein degradation.
Aging and ER stress in muscle
Sarcopenia is an important clinical problem. Despite it has high prevalence, no clear understanding has been developed. Sarcopenia is associated with multiple factors, including increased pro-inflammation cytokine, oxidative stress, ER stress and fat infiltration as well as reduced mitochondrial function. Intramyocellular lipid (IMCL) accumulation in skeletal muscle was suggested to play a role in the development of sarcopenia with aging. IMCL associated with insulin resistance by increased mitochondrial dysfunction in elderly individuals. Aging has been shown to be accompanied by numerous functional alterations of mitochondrial such as increased ROS. Increased ROS due to imbalance of redox environment can promote protein breakdown directing muscle fibers into a catabolic state that ultimately leads to muscle wasting. Furtherover, increased oxidative stress due to ROS closely links with increase insulin resistance, inflammation and ER stress in skeletal muscle [39].
Previous studies proposed that perturbation in cellular proteostasis is one of the crucial hallmarks of aging [40]. Proteostasis is regulated by network of mechanisms related to protein synthesis, folding, trafficking, secretion, and degradation [41]. The disruption of proteostasis network was observed in obesity and aging patient that cause imbalance between anabolism and catabolism related to muscle wasting. The key players for proteostasis network include work include chaperones and foldases, UPR, the mitochondrial UPR, ERAD machinery, autophagy pathway, the ubiquitin–proteasome system, heat-shock response, the integrated stress response, and the mechanisms controlling redox balance. Both of UPR and autophagy pathway are tightly coordinated to control protein quality. Autophagy is a catabolic process that is necessary for removal of aggregate protein and abnormal organelles in a lysosomal-dependent manner, therefore, excess of autophagy results in the loss of muscle mass, whereas reduced of autophagy causes degeneration and weakness of skeletal fiber [42]. Indeed, muscle specific knockout of the autophagy genes Atg5 or Atg7 in mice resulted in an increase muscular atrophy. Taken together, we speculate that interaction between UPR and autophagy may play an important role to maintain the muscle protein quality control.
The key factor in protein synthesis pathway for protein quality control is mammalian target of rapamycin complex 1 (mTORC1), indeed aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis [43]. Furthermore, high fat diet leads to the development of insulin resistance, in part, by decreased Insulin/IRS-1/PI3-K/Akt signaling pathway. Since insulin plays an important role in the initiation of protein synthesis through Akt/mTOR pathway, decreased activation of Akt/mTOR pathway by high fat diet and aging may induce muscle atrophy due to imbalance between protein synthesis and degradation. Indeed, mTORC1 by insulin growth factor or amino acid act to inhibit autophagy. Although the mechanism is not clear, mTORC1 activity is probably regulated in part through feedback loop to prevent insufficient of excessive autophagy, that is may linked with protein quality control in skeletal muscle.
ER stress and Autophagy by Exercise
Exercise training is considered an effective instrumental to prevent and decrease risk factor of insulin resistance, metabolic dysfunction, and muscle atrophy [44]. Induction of oxidative stress, mitochondrial dysfunction, ER stress and pro-inflammatory cytokines were common marker of insulin resistance, T2DM and muscle atrophy in skeletal muscle. Exercise training provides the multiple positive effects on metabolic dysfunction and serves to maintain the muscle function and quality via an increase antioxidant and mitochondrial oxidative function and a reduction of inflammatory cytokines expression. Increased oxidative stress, mitochondrial dysfunction and inflammatory cytokines were associated with ER stress. However, information of exercise training effects on ER stress link with skeletal muscle dysfunction involving insulin resistance, T2DM, muscle atrophy is not enough to understand.
Induction of UPR has been found in skeletal muscle of myophthies patients, in mice but not humans fed a high fat diet [36], and with a combination of bed rest and exogenous insulin administration [45]. However, despite the role of UPR by ER stress in skeletal muscle is important to maintain muscle function, there is not enough studies on exercise related to UPR has been done to understand ER stress in skeletal muscle, especially, how UPR contributes to adapt skeletal muscle function by exercise training. Transcription coactivator peroxisome proliferator-activator receptor gamma coactivator-1 alpha (PGC-1α), which is a key regulator of mitochondrial function, oxidative metabolism, and energy homeostasis in skeletal muscle [46], exercise increases PGC-1α, BiP, ERdj4, GADD34, ATF3, ATF4, CHOP and XBP-1S gene, which is an UPR marker gene. In addition, PGC-1α overexpression resulted in an increase BiP, GRP94, ATF3 and CHOP, and this study also has been demonstrated that ATF6α is required for PGC-1α regulated induction of UPR gene using ATF6α knock out mice, indicating that PGC-1α by exercise interacts with ATF6α to trigger transcription of UPR gene [47]. In human exercise study, ultra-endurance exercise has been shown to increase the UPR activation such as ATF6, IRE1, BiP/GRP78 and spliced XBP1 mRNA [48]. Furthermore, ATF6 and IRE1α have also been observed in human skeletal muscle following acute resistance exercise, whereas PERK, eIF2v and CHOP were not increased [49]. Resistance exercise also increases PGC-1α [50], indicating that both of acute aerobic and resistance exercise increase UPR through PGC-1α. Interestingly, these results indicate that differential UPR pathway by ER stress might be triggered by different by exercise type such as endurance and resistance.
Rutkowski and Hegde [51] have suggested that the UPR is as adaptive response pathway to maintain physiological function to defend cell and tissues against foreign insults and pathological events. Wu et al. [47] also showed that ER chaperones, BiP and GRP94, and the co-chaperone, ERdj4, were increased after treadmill run in both acute and trained muscle. Interestingly, a stress marker, ATF3, was only induced by 3-fold in the training group compared to almost 20-fold in mice only run once. Furthermore, CHOP and spliced form XBP-1 mRNA were not changed while being significantly elevated in the run once mice. Furthermore, ATF4 was lower in trained mice when compared to sedentary control mice, indicating that physiological ER stress in skeletal muscle due to moderate exercise may lead to adaptation and protect skeletal muscle against further stress [47]. In fact, it is well known that PGC-1α is closely associated with numerous factor in muscle adaptation and function including regulation of mitochondrial function, redox environment, pro-inflammatory cytokine and muscle mass by exercise training. Thus, further studies are necessary to understand how UPR by ER stress can mediate skeletal muscle adaptation by exercise training, and can improve health benefit in variety negative environmental on muscle.
ER stress is also connected with autophagy which is tool for both protein and organelle quality control, the benefit of an increased autophagy is linked with a life extending key mechanism for calorie restriction and calorie restriction mimetics in different species [52]. Moreover, autophagy protects against diseases, including inflammatory diseases, ageing, insulin resistance and cancer [53]. Since autophagy is directly linked with mitochondria, and mitochondrial dysfunction can lead to ER stress, exercise, thus, can reduce ER stress by acting upon mitochondrial protein homeostasis. Indeed, endurance exercise stimulates autophagy in skeletal muscle and heart [54-56], and the autophagy is required for exercise training-induced skeletal muscle adaptation and in improvement of physical performance in lean mice [57], as such it is necessary for exercise-mediated preservation of insulin sensitivity in muscle of obese mice [55].
Bcl-2 act as an anti-apoptotic and anti-autophagy protein that inhibits autophagy program via a direct interaction with autophagy protein beclin-1 at ER [58]. The autophagy induced by exercise has shown to involve the disruption of Bcl-2/beclin-1 complex [55]. Disruption of the Bcl-2/Beclin-1 complex induces the autophagy in mammalian cells [58]. Interestingly, Bcl-2 dysfunction mice resulted in a decrease endurance and altered glucose metabolism during acute exercise, as well as in an impairment of chronic exercise-mediated protection against high fat diet induced glucose intolerance [55]. Therefore, Bcl-2 induced by exercise regulate autophagy that is required for muscle glucose homeostasis [55].
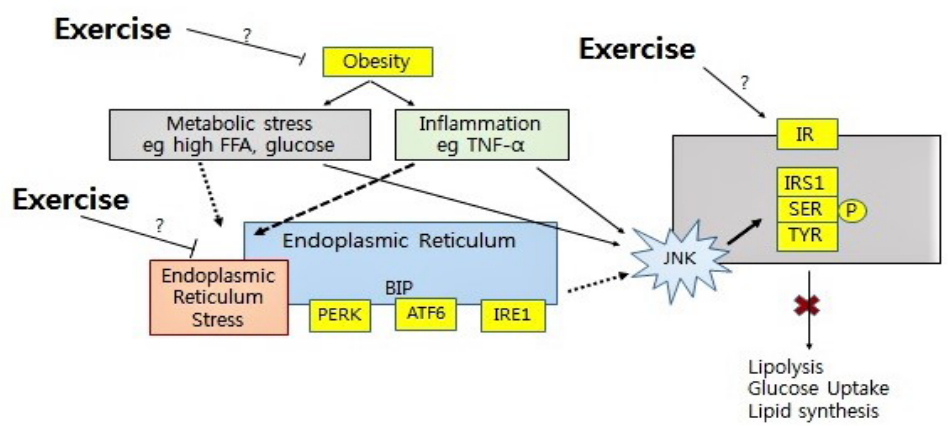
The autophagy has emerged as an important player to protect against development of insulin resistance and T2DM [59], and to maintain mitochondrial function and skeletal muscle mass (Figure 1) [60]. Particularly, elimination of dysfunctional mitochondria by mitophagy, a form of macroautophagy selective for mitochondria, is important for muscle energy.
It seems that the differential response of autophagy and ER stress according to exercise phase such during or after exercise. During exercise, mTOR pathway might be decreased that may result in an increase autophagy, because mTOR is a negative feedback factor for autophagy. Particularly, increased mTOR after exercise will be decrease autophagy. Acute exercise increases UPR by PGC-1α/ATF6 complex in skeletal muscle, however, differential UPR responses by exercise in trained individuals, indicating that repetitive exercise training induces the adaptive arm of the UPR and protects skeletal muscle against further stress caused by future exercise [47].
Conclusions
The greatest challenges in an aging society include the health and financial stability of seniors. Many recent studies of comestibles and medicines have focused on improving the health of seniors. However, the effectiveness of this research is unclear, particularly considering the financial decline in Korean seniors. Exercise can fundamentally prevent and relieve symptoms of sarcopenic obesity, which is primarily caused by a decline in physical activity; this is a financially effective method. However, despite the potential benefits, implementation in seniors is very low. Issues include the insufficient scientific basis for the effect of exercise training on sarcopenic obesity and the absence of a detailed treatment program (type, intensity, time, frequency, etc.). Therefore, it is necessary to establish the mechanism of insulin resistance syndrome, with a focus on sarcopenic obesity and ER stress in obesity and the process of aging, and to clarify the precise effects of exercise training from a molecular biological perspective to prevent and delay aging-related changes.